The ambitious package of measures
The European Commission has proposed an ambitious package of measures to improve the health of EU citizens while ensuring the long-term resilience and competitiveness of the healthcare sector. This comprehensive initiative includes a Biotech Act, revised rules for medical devices, and a Safe Hearts Plan which is commonly referred to as the “Omnibus” regulation. Since that is quite a lot to take in, we are going to unpack exactly how these changes will simplify the road ahead.
This sweeping legislative initiative acts as a bridge between multiple regulatory silos to create a more modern and efficient ecosystem. The broader package includes the landmark Biotech Act to build a world leading biotechnology industry along with the “Safe Hearts Plan“ which is a comprehensive strategy to tackle cardiovascular disease as the leading cause of death in Europe. Furthermore, it overhauls medical device regulations to transform the EU into a more agile environment for health technology while ensuring that the path from laboratory to patient is both faster and more efficient.

Medical devices
Despite the EU market leadership in the field of medical devices, worth 170 billion euros and a workforce of nearly one million people, current regulatory bottlenecks are causing significant delays and rising costs. This reform addresses these challenges by digitizing procedures, establishing clear assessment timelines, and expanding the mandate of the EMA to offer technical expertise and manage device shortages. By creating a coherent framework for AI enabled devices, the EU expects to achieve 3.3 billion euros in annual savings while ensuring the highest standards of safety and global competitiveness.
Within this massive legislative effort, the focus specifically sharpens on the Omnibus MDR and IVDR proposal which fundamentally redefines how AI driven medical technologies are governed. For developers, the “Omnibus” nature of this reform means that the AI Act, Medical Devices Regulation (MDR), and In Vitro Diagnostics Regulation (IVDR) will finally operate under a single and coherent framework. By introducing uniform rules for devices incorporating AI and strengthening the role of the European Medicines Agency (EMA), the proposal eliminates the legal uncertainty and administrative bottlenecks that have delayed innovation. This targeted reform ensures that patient safety remains the highest priority. The path from the laboratory to the market is streamlined to offer a predictable and digital first environment for the next generation of healthcare technology.
Strategic Pillars of the Medical Device Reform
In the context of the new Omnibus proposal, medical devices ranging from instruments and implants to software and reagents are defined as essential tools for the diagnosis, prevention, and treatment of disease or injury. The following pillars outline how this sweeping reform simplifies the journey for these technologies from the laboratory to the patient.
Regulatory and Administrative Efficiency

The reform significantly reduces the administrative burden on manufacturers. The costly 5-year maximum validity period for notified body certificates is abolished and replaced with continuous, risk-proportionate periodic reviews.
For low- and medium-risk devices (such as class IIa, non-implantable class IIb, and class C IVDs), notified bodies will only need to assess the technical documentation of one representative device per generic group or category. The rules also introduce a framework for “well-established technology devices,” exempting them from certain stringent clinical and conformity requirements.
Furthermore, AI-powered devices will no longer face redundant assessments, as the application of the AI Act will be limited to prevent a double layer of regulatory requirements. Finally, reporting duties are lightened by reducing the frequency of Periodic Safety Update Reports (PSURs) and removing the need for notified bodies to separately validate the draft Summary of Safety and Clinical Performance (SSCP).
Innovation and Competitiveness
To boost the EU’s global competitiveness, the proposal establishes adaptive pathways for “breakthrough devices” and “orphan devices“. Once designated by an expert panel, these devices benefit from prioritized assessments, rolling reviews, and conditional market access based on limited pre-market clinical data, provided the manufacturer commits to post-market follow-up.
The legislation also introduces national and Union-level “regulatory sandboxes” allowing developers to test innovative technologies in controlled environments under regulatory supervision. To reduce reliance on animal testing and lengthy trials, the definition of clinical evidence is expanded to promote New Approach Methodologies (NAMs), such as in silico testing and computer modeling.

Patient Safety and Product Availability

To prevent shortages of critical devices, the proposal mandates a centralized IT tool within Eudamed for manufacturers to report supply interruptions. The EMA will develop a methodology to identify and publish a list of critical devices.
The rules also include a “grandfathering” clause for legacy orphan devices, allowing them to remain on the market beyond transitional periods without a new conformity assessment if they meet specific safety criteria.
In the event of public health emergencies or crises, the Commission and Member States gain new powers to authorize the distribution of critical devices even if standard conformity assessments are incomplete.
Coordination and Cooperation
The governance structure is overhauled to ensure harmonized practices across the EU. The coordination mechanism (the “Helsinki procedure”) is codified to resolve disputes between Member States over product classification, utilizing expert panels for guidance.
The EMA’s role is expanded to provide scientific, technical, and administrative support to national authorities for vigilance, market surveillance, multi-country clinical studies, and borderline classifications.
The oversight of notified bodies is streamlined through Joint Assessment Teams, and the requirement for a full reassessment every five years is eliminated. Additionally, the proposal promotes global convergence by officially supporting international reliance mechanisms like the International Medical Device Regulators Forum.

Digitalisation

The proposal transitions compliance into a digital-first environment. Manufacturers can provide the EU declaration of conformity electronically, and subject to future implementing rules, product labels and instructions for use may also be provided in digital formats.
Technical documentation submitted to notified bodies can now be shared in structured, machine-readable formats to easily enable retrospective conformity checks.
To close cybersecurity gaps, manufacturers of connected devices must report actively exploited vulnerabilities and severe cybersecurity incidents directly to national Computer Security Incident Response Teams (CSIRTs) and ENISA via Eudamed.
Support for Small and Medium-Sized Enterprises (SMEs)
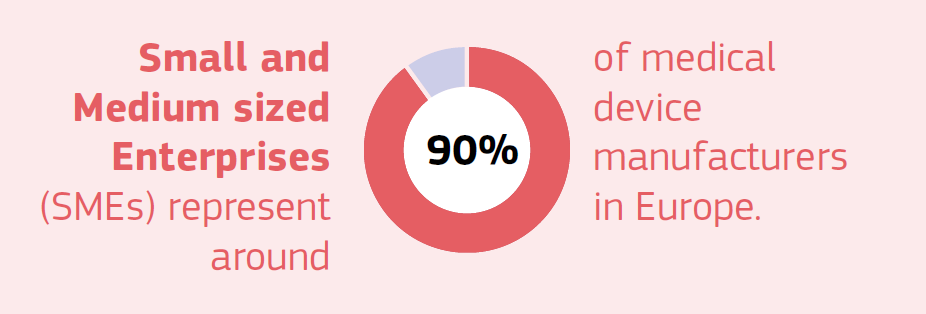
Since SMEs make up roughly 90% of the medical technology industry, the proposal specifically alleviates their regulatory burdens. Micro and small enterprises relying on an external Person Responsible for Regulatory Compliance are no longer required to have them “permanently and continuously” available.
Notified bodies are legally required to offer a 50% fee reduction for micro-enterprises and a 25% reduction for small enterprises, alongside the option to defer fee payments until the assessment is finalized.
Finally, the EMA is mandated to establish a dedicated support scheme to offer regulatory guidance specifically tailored to SMEs.
If you are looking for a deeper dive into these regulatory changes, you can explore the detailed Questions and Answers: Medical Devices document for specific technical clarifications. For a high-level visual summary of the reform’s goals and its expected impact on the industry, the Factsheet: Medical Devices is an excellent additional resource. Both documents are part of the Commission’s official package to help manufacturers and developers navigate this new era of innovation.
Sources
- European Commission (16 December 2025). Proposal for a Regulation of the European Parliament and of the Council amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards simplifying and reducing the burden of the rules on medical devices and in vitro diagnostic medical devices (COM(2025) 1023 final, 2025/0404 COD). Strasbourg
- European Commission (16 December 2025). Annexes 1 to 2 to the Proposal for a Regulation of the European Parliament and of the Council amending Regulations (EU) 2017/745 and (EU) 2017/746 (COM(2025) 1023 final). Strasbourg
- European Commission (16 December 2025). Questions and answers on simpler and more effective rules for medical devices. Brussels